
Research
I’m working at Microsoft Research as a researcher on DeepQMC for electronic structure. I graduated with Prof. Thomas F. Miller III @ CCE, Caltech in 2022 on AI for quantum chemistry (MOB-ML). Before joining Microsoft Research AI4Science lab, I was a researcher working on the interdisplinary studies between quantum sciences & machine learning in Tencent Quantum Lab for around 9 months. The topics/projects I am working/worked on are listed as below.

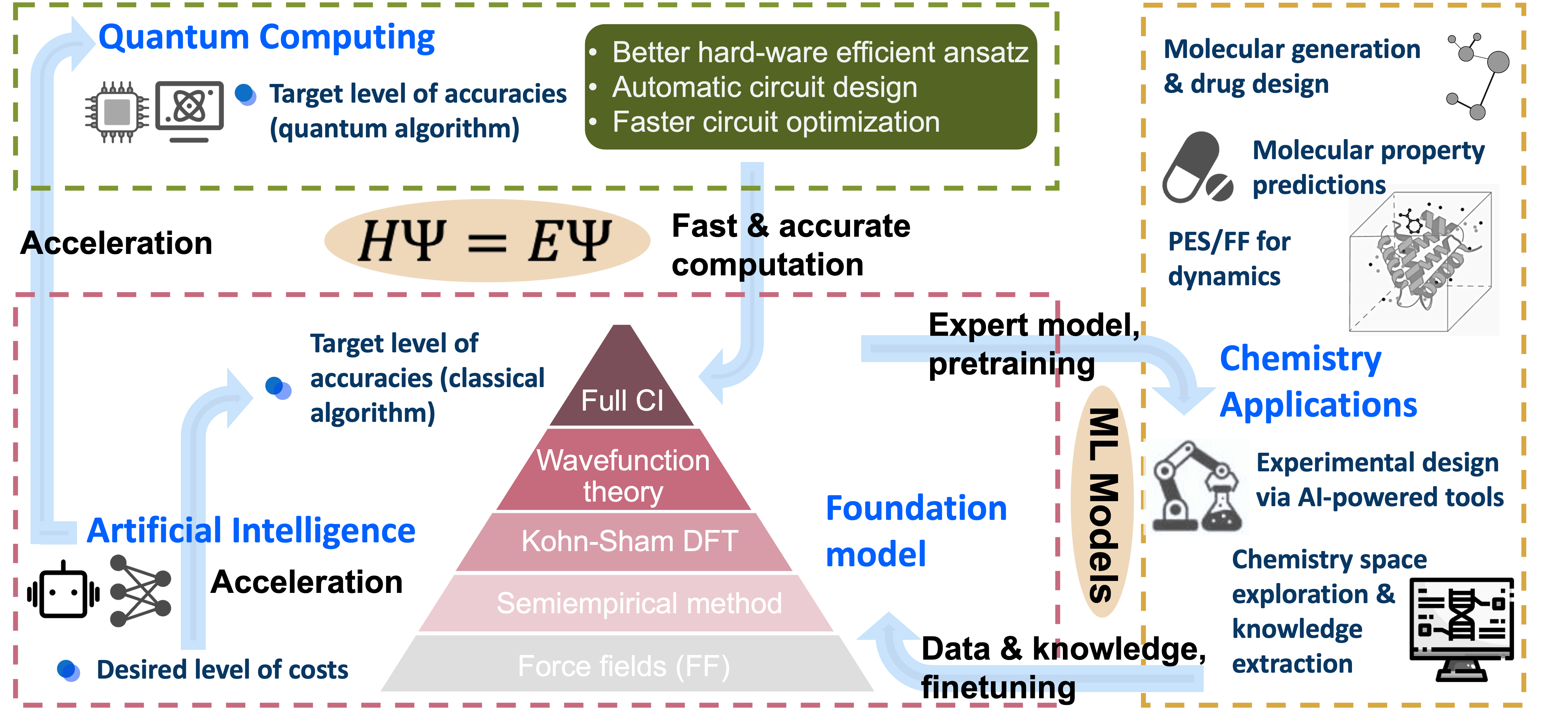
Microsoft Research work:
AI for chemistry
Deep Quantum Monte Carlo for Electronic Structure
[1] Cheng, L.*; Szabó, P.B.*; Schätzle, Z.*; Kooi, D.; Köhler, J.; Noé, F.; Gori-Giorgi, P.; Foster, A. Highly Accurate Real-space Electron Densities with Neural Networks, arXiv:2409.01306.(*co-first author)Link
LLMs for Scientific Discovery: Evaluate and explore the ability of LLMs in many scientific tasks.
[1] MR AI4Science^, MA Quantum. The Impact of Large Language Models on Scientific Discovery: a Preliminary Study using GPT-4. arXiv:2311.07361 (2023).Link (^Main contributor to Chapter 4, see Authorship and contribution list)
Other independent work:
AI for quantum computing & Quantum machine learning:
The goal is to apply the state-of-the-art ML tools to facilitate more efficient quantum algorithm realizations and quantum resource usages.
[1] Cheng, L.*; Chen, Y.-Q.*; Zhang, S.-X.; Zhang, S. Error-mitigated quantum approximate optimization via learning-based adaptive optimization. (*co-first author) arXiv:2303.14877 (2023).Link
AI for biology: Bayesian Optimization for experimental design
[1] Cheng, L.*; Yang, Z*; Liao, B; Hsieh, C; Zhang, S. ODBO: Bayesian optimization with search space prescreening for directed protein evolution. Link
Quantum computing for quantum chemistry
[1] Sun, J.; Cheng, L.; Li, W. Towards chemical accuracy with shallow quantum circuits: A Clifford-based Hamiltonian engineering approach. J. Chem. Theory Comput., 2024.Link
[2] Li, W; Allcock, J.; Cheng, L.; Zhang, S.-X.; Chen, Y.-Q.; Mailoa, J.P.; Zhang, S. TenCirChem: An efficient quantum computational chemistry package for the NISQ era. J. Chem. Theory Comput., 2023.Link
Caltech work:
AI for electronic structure: MOB-ML
 Using information from Hartree-Fock thoery and molecular oribtal based representation to learn ground state molecular properties. More upcoming research studies on this topic, please refer to my group page
Using information from Hartree-Fock thoery and molecular oribtal based representation to learn ground state molecular properties. More upcoming research studies on this topic, please refer to my group page
Selected publications:
[1] Cheng, L.; Sun, J.; Deustua, J. E.; Bhethanabotla, V. C.; Miller III, T. F. Molecular-orbital-based machine learning for open-shell and multi-reference systems with kernel addition Gaussian process regression. J. Chem. Phys., 2022. Link
[2] Lu, F.*; Cheng, L.*; DiRisio, R. J.*; Finney, J. M.; Boyer, M. A.; Lu, F.; Moonkaen, P.; Sun, J.; Lee, S. J. R.; Deustua, J. E.; Miller III, T. F.; McCoy, A. B. Fast near ab initio potential energy surfaces using machine learning. J. Phys. Chem. A, 2022. (*co-first author) Link
[3] Cheng, L.; Kovachki, N; Welborn, M.; Miller III, T. F. Regression clustering for improved accuracy and training costs with molecular-orbital-based machine learning. J. Chem. Theory Comput., 2019. Link
[4] Cheng, L.; Welborn, M.; Miller III, T. F. A universal density matrix functional from molecular orbital-based machine learning: Transferability across organic molecules. J. Chem. Phys., 2019. Link
[5] Welborn, M.; Cheng, L.; Miller III, T. F. Transferability in machine learning for electronic structure via the molecular orbital basis. J. Chem. Theory Comput. 2018. Link (Highlighted with commentary in C&EN and Caltech News)
AI for biology: INSPIRE
Predict the thermaldynamics properties of nucleic acids using their secondary structures.